太倉中國eCTD常用解決方案
賦悅Word插件自主研發Word插件kuai速編輯:整合word常用功能按鈕,避免頻繁切換菜單;內置標題、段落、文字、目錄、超鏈接等的格式和樣式,可kuai速設置和更文檔的格式kuai速鏈接:雙擊或者拖拽的方式,制作文本超鏈接或者題注超鏈接;可搜索全文關鍵字,自動制作超鏈接文檔拆分:可根據不同的條件將word文件顆粒化,如分節符、頁眉、頁腳、頁碼范圍和自定義頁碼等PDF轉換:WORD轉PDF,自動判斷是否生成書簽,自動鑲嵌所有字體,生成PDFkuai速網頁瀏覽的PDF,確保生成的PDF所有屬性符合法規要求文檔驗證:驗證文檔的字體、字號、紙張、頁面布局、空白頁、頁碼、編號、目錄、超鏈接等,并且可以ding位驗證結果可定制:可根據用戶需求定制格式和樣式模板。 歐盟DMF注冊申報相關技術支持。太倉中國eCTD常用解決方案

電子簽章與傳輸安全文件需經AES-256加密后刻錄至不可擦寫光盤,并附MD5校驗碼。光盤損壞或bing毒污染將觸發重遞交流程,原載體按銷毀程序處理。審評與核查協同自2018年起,FDA要求提交兩套光盤分別用于審評和現場核查,2022年調整為“1套審評+1套核查+1套專項資料”模式,提升流程效率。guo際化兼容性增強美國eCTD系統支持與歐盟、日本等地區的XML互操作性,但區域差異(如模塊1的標簽要求)仍需人工適配。未來通道創FDA計劃引入API接口支持企業系統直連,并探索基于云存儲的實時提交與審評,減少物理媒介依賴。 無錫賦悅科技eCTD報價中NDA注冊申報相關技術支持。
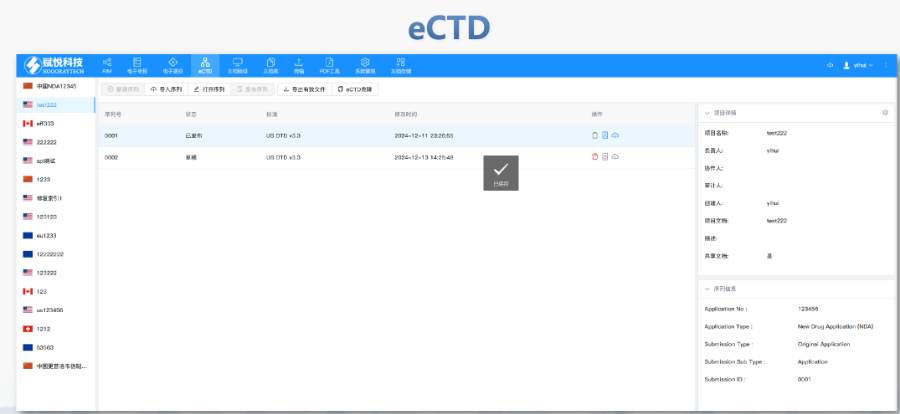
2015年發布《關于yao品醫療器械審評審批制度的意見》,提出yao監五大目標,將eCTD納入guo家yao監數字化戰略。2017年,中guo加入ICH(guo際人用yao品注冊技術協調會),成為全球第八個監管機構成員,加速與guo際標準接軌。2018年,guo家yao監局(NMPA)完成eCTD文檔管理系統招標,由上海寶信與德國LORENZ合作搭建技術平臺,標志著技術基礎設施的落地。規范制定與試點階段(2019-2023年)2019-2020年,CDE(yao品審評中心)發布《eCTD技術規范》《驗證標準》等征求意見稿,并zu織兩輪企業測試。2021年,NMPA明確化學yao1類、。2022年實施電子申報(非eCTD格式),2023年取消紙質資料提交,為eCTD鋪開奠定基礎。實施與擴展階段(2024-2025年)2024年3月更電子申報技術要求,7月啟動網絡傳輸試點。2025年1月27日,NMPA將eCTD適用范圍擴大至化yao1-5類臨床試驗及上市申請、生wu制品1-3類全流程,覆蓋yao、仿制yao及生wu類似yao,實現與guo際主流申報模式同步。
仿制yao作為提高yao物可及性與可負擔性的一類yao物,2012年以前,注冊審評是不收取任何費用的,但當時仿制yao申請積壓嚴重,從申報到獲批需要3~5年的時間。美國國會于2012年頒布了仿制yao使用者費用修正案(GenericDrugUserFeeAmendments,GDUFA),該法律要求制yao行業支付一定的用戶費用,以補充仿制yao申請的審評以及現場檢查的費用,減少仿制藥申請積壓,縮短審評時間,增加基于風險的現場檢查等,其目的是加快公眾獲得安全you效的仿制yao,并降低行業成本。GDUFA必須每五年重授權一次,于2017年更(GDUFAII),于2022年更(GDUFAIII);目前收費種類分為以下四種:ANDA審評費、DMF審評費,在審評時一次性繳納;項目費(Programfee)、設施費(Facilityfee),是上市后每年繳納一次。 加拿大eCTD驗證標準相關技術支持。
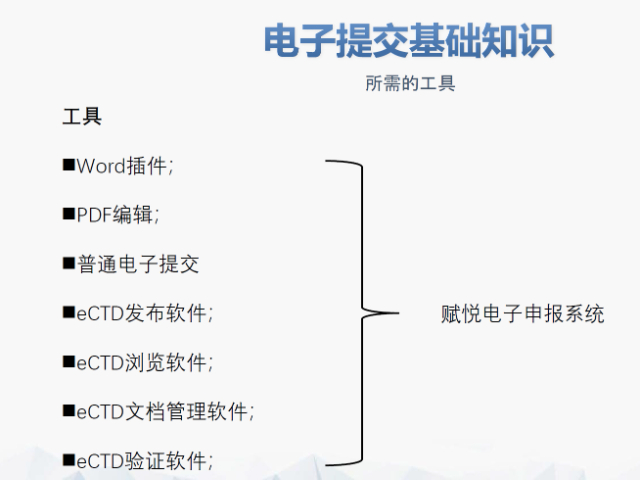
eCTD的實施為監管機構和企業帶來了多重機遇。電子化申報資料能夠極大地加速審評效率,減少人為判斷錯誤和數據混淆的情況,從而提高審評的準確性和速度。同時,eCTD帶來的數據標準化機遇使得全球監管機構的資料內容和電子格式得以統一,有助于在不同監管機構之間進行數據傳輸和共享。這對于提升全球監管效率和行業研發效率具有重要意義。此外,eCTD的實施還促進了全球合作,構建了全球監管的底層大數據基礎。對于企業而言,eCTD提供了一個規范化的研發活動模板,有助于降低與監管機構溝通的成本,提高申報效率。特別是對于國內的醫技術企業而言,eCTD的實施更是具有重要意義,有助于這些企業更好地走向全球市場。然而,中小企業在享受這些機遇的同時,也面臨著技術和成本壓力。eCTD的實施需要專門的團隊進行系統維護和開發,這對于中小企業來說是一筆不小的開支。同時,數據安全問題也是企業關注的焦點。此次CDE擴大eCTD實施范圍對行業而言是一個積極的風向標。短期內,企業面臨的挑戰包括適應更高要求的技術規范并提高文件質量、和eCTD出版系統的磨合以及進行eCTD知識的跨職能培訓等。 中ANDA注冊申報相關技術支持。山東仿制藥eCTD遞交
加拿大NDA注冊申報相關技術支持。太倉中國eCTD常用解決方案
美國藥物主文件(Drug Master File, DMF)是向FDA提交的機密技術文件,用于支持藥品生產、質量控制及合規性審查。以下為申報的要點和流程總結: DMF概述與類型 定義與作用 DMF是藥品生產全過程的詳細檔案,包含原料藥、輔料、包裝材料等的生產設施、工藝、質量控制等信息,供制劑廠商引用以支持其注冊申請。其意義在于保護企業機密的同時,滿足FDA對供應鏈透明度的要求。 DMF類型 Ⅱ類:原料藥、中間體及制劑(如微生物外泌體、細胞株等生物制品均屬此類)。 Ⅲ類:包裝材料。 Ⅳ類:輔料、著色劑等添加劑。 Ⅴ類:非臨床/臨床數據等特殊信息(需FDA預先批準)。 注:Ⅰ型(生產設施與人員)已于2000年停用。太倉中國eCTD常用解決方案
- 遼寧仿制藥eCTD 2025-12-19
- 浦東新區仿制藥eCTD服務電話 2025-12-19
- 無錫賦悅科技eCTD報價 2025-12-19
- 上海國產eCTD服務電話 2025-12-19
- 河北國際注冊eCTD 2025-12-19
- 吳江區賦悅科技eCTD使用 2025-12-19
- 靜安區仿制藥eCTD哪個品牌好 2025-12-19
- 杭州國際注冊eCTD歡迎選購 2025-12-19
- 太倉中國eCTD常用解決方案 2025-12-19
- 閔行區NDAeCTD便宜 2025-12-19
- TO封裝技術廠家 2025-12-19
- 山西一維無線掃描槍 2025-12-19
- 鹽田區新型智能手表 2025-12-19
- 湖南移動電源3C認證哪家便宜 2025-12-19
- 湛江小型民宿管理系統哪種好 2025-12-19
- 智能終端 2025-12-19
- 臨沂一對多掃描槍推薦 2025-12-19
- 紹興晶圓切割代工廠 2025-12-19
- 遼寧仿制藥eCTD 2025-12-19
- 什么是打印紙哪家便宜 2025-12-19